Mengenal Sindrom Marfan

SM atau Sindrom Marfan adalah kelainan genetik yang mempengaruhi jaringan konektif (ikat) dalam tubuh. Marfan syndrome sangat jarang terjadi, mempengaruhi sekitar 1 dari 5.000 orang dan biasanya menderita bertubuh sangat tinggi serta ramping. Bahkan jika seseorang terlahir dengan marfan syndrome, mereka mungkin baru mengetahuinya di kemudian hari. Tidak ada obat untuk sindrom ini.
Prevalensinya sekitar 2-3 per 10.000 penduduk dan sekitar 25-30?alah suatu mutasi baru. Berdasarkan data catatan medis Pusat Jantung Nasional Harapan Kita (PJNHK), tercatat 39 kasus pada tahun 2006-2012 dan 6 kasus diantaranya dilakukan operasi katup aorta.
Sindrom Marfan disebabkan oleh mutasi atau kelainan pada gen FBN1. Normalnya, gen ini berfungsi untuk memproduksi protein fibrilin, yang tugasnya adalah membangun jaringan ikat elastis pada tubuh dan mengontrol pertumbuhan. Mutasi pada gen ini membatasi kemampuan tubuh untuk memproduksi fibrilin. Akibatnya, pertumbuhan jaringan ikat elastis menurun dan pertumbuhan tulang jadi tidak terkontrol.
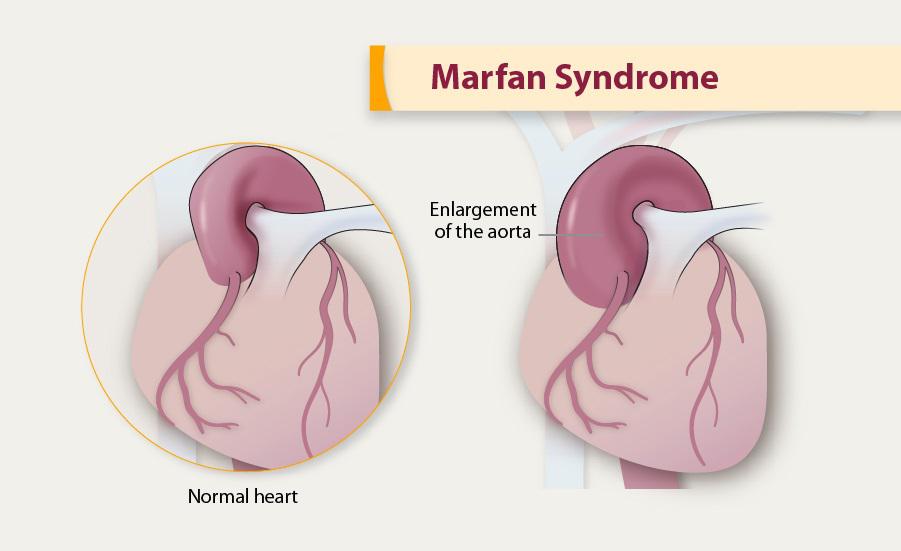
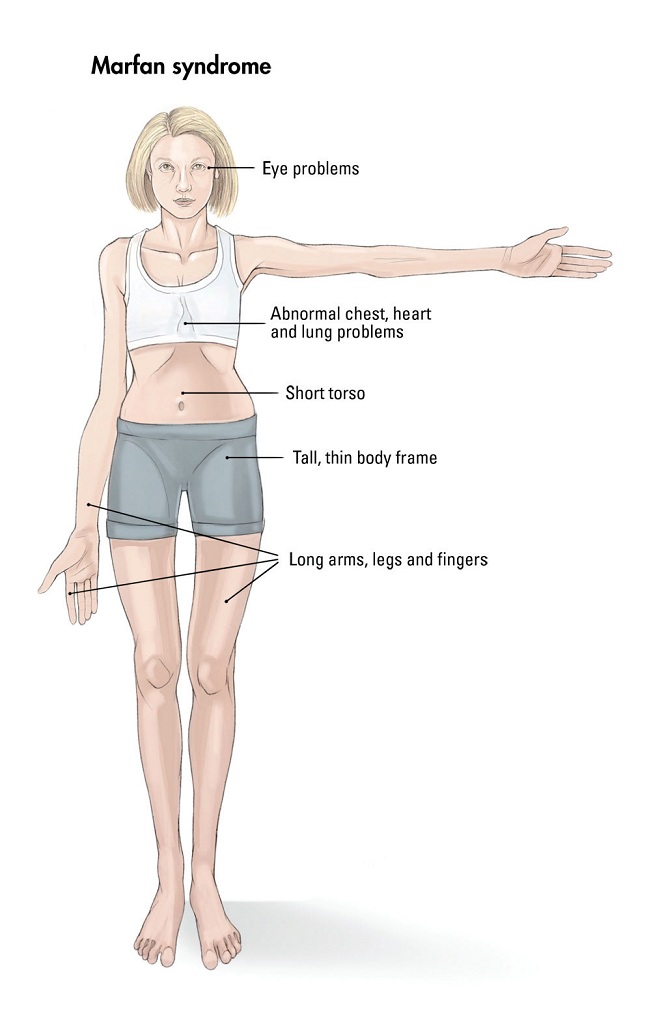
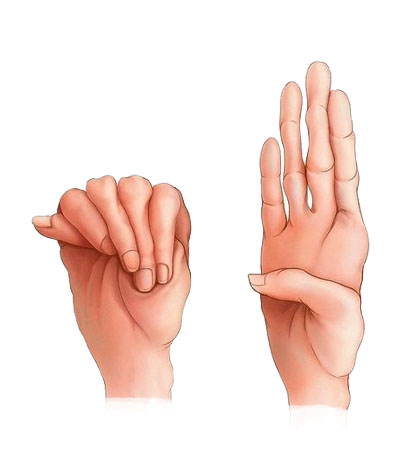
Bayi dan anak yang mengidap Marfan syndrome biasanya berperawakan kurus dan tinggi. Lengan, kaki, serta jari-jarinya mungkin terlihat seperti tidak proporsional dan terlalu panjang untuk ukuran tubuhnya. Selain itu, tulang punggung anak dengan sindrom ini mungkin terlihat bengkok sehingga ada beberapa penderitanya yang juga mengidap kyphoscoliosis. Sendi-sendi bayi dan anak dengan Marfan syndrome juga terasa lemah dan mudah bergeser. Umumnya, wajah pengidap sindrom Marfan berbentuk panjang dan tajam, disertai dengan bentuk langit-langit mulut yang lebih tinggi dari orang normal. Bayi dan anak dengan sindrom ini juga biasanya memiliki masalah pada gigi dan langit-langit mulut. Di samping itu, tulang pada bayi dan anak yang mengalami sindrom Marfan umumnya bermasalah seperti bentuk tulang telapak kaki yang rata, adanya hernia, dan pergeseran tulang. Gejala lainnya yang dapat dilihat pada bayi dan anak dengan Marfan syndrome ini adalah masalah pada penglihatan. Sindrom ini menyebabkan penglihatan bayi menjadi lebih buram, jarak pandang lebih dekat, lensa mata bergeser, atau ada perbedaan bentuk antara mata kiri dan kanan. Sebanyak 90?yi dan anak dengan Marfan syndrome mengalami perubahan pada fungsi jantung dan pembuluh darah. Sindrom marfan ini dapat merusak dinding pembuluh darah. Hal tersebut berpotensi memicu terjadinya aneurisma aorta, diseksi aorta, atau pecahnya aorta. Bayi dan anak dengan Marfan syndrome juga kemungkinan dapat mengalami perdarahan di dalam tengkorak kepala atau aneurisma otak. Bayi dan anak dengan Marfan syndrome ini juga berisiko mengalami masalah pada katup jantungnya, seperti prolaps katup mitral. Hal ini biasanya ditunjukkan dengan gejala napas pendek, tubuh sangat lelah, atau detak jantung tidak beraturan.
Tidak ada cara untuk mencegah SM, pasien dan kerabat mereka mungkin ingin mencari konseling genetik untuk membicarakan risiko mereka menularkan kelainan tersebut kepada anak-anak mereka. Seseorang dengan SM memiliki risiko 50% untuk meneruskan gen abnormal tersebut kepada anak. Penting untuk diingat bahwa diagnosis dan pengobatan dini mencegah perkembangan komplikasi yang jauh lebih serius. Terlepas dari risiko tinggi yang terkait dengan masalah kardiovaskular, orang dengan SM memiliki harapan hidup rata-rata sekitar 70 tahun.
Referensi :
NORD: Marfan Syndrome: https://rarediseases.org/rare-diseases/marfan-syndrome/
Harvard Health: Marfan Syndrome: https://www.health.harvard.edu/a_to_z/marfan-syndrome-a-to-z
https://ibupediacdn.imgix.net/images/7hkf0m4e9xpknhowu.jpg
https://koran-jakarta.com/images/library/madsrR3.jpg
https://d324bm9stwnv8c.cloudfront.net/Gejala_dari_Sindrom_Marfan_yang_Wajib_Diwaspadai.jpg
https://gayasehatku.com/wp-content/uploads/2022/03/MARFAN-SYNDROME-HAND.jpg







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}